


ALS Pathology Linked to Defects in the Cell Nucleus

Packard researchers used Answer ALS cell lines to show that the protein CHMP7 accumulates in the nucleus, triggering a series of events that creates some of the molecular hallmarks found in both sporadic and familial as well as FTD.
Pathological changes in the cellular localization of a protein that helps maintain the nuclear pore (large protein complexes that move macromolecules between the nucleus and cytoplasm) is an early step in the development of ALS and FTD, according to a new study in Science Translational Medicine. Packard Center founder and director Jeffrey Rothstein MD, PhD, a neurologist at Johns Hopkins University, his postdoctoral fellow Alyssa Coyne PhD, and colleagues showed that the protein CHMP7 accumulates in the nucleus, triggering a series of events that creates some of the molecular hallmarks found in both sporadic and familial ALS as well as FTD, including nuclear pore dysfunction and TDP43 loss of function and cytoplasmic accumulation.
“This work identifies one of the earliest events in not only familial ALS but also the far more common sporadic ALS. The use of patient derived cell lines is like a biopsy used in cancer research. It has really allowed us to make significant progress towards understanding how ALS occurs in the absence of known genetic mutations,” Coyne says.
Although scientists have learned a lot about the end stages of ALS, they know far less about the molecular events that ultimately spark the cascade that results in motor neuron degeneration. Making this work even more challenging is the underling heterogeneity of ALS/FTD. Nine out of 10 cases are sporadic and linked to no known genetic mutations. Despite these underlying differences, 97% of ALS cases clear a DNA and RNA-binding protein called TDP43 from the nucleus and accumulate it in the cytoplasm, resulting in a loss of nuclear function. Newer work also shows changes to the nuclear pore. But if and how these two alterations were linked wasn’t clear, nor were the processes that triggered these issues.
In the current study, Coyne and colleagues showed that induced pluripotent stem cell-derived motor neurons from patients with sporadic ALS showed some of the same nuclear pore alterations as those seen in C9orf72-ALS/FTD. In particular, these cells showed marked reductions in specific nucleoporins (Nups), proteins that comprise the nuclear pore complex. One of the proteins that helps to regulate the integrity of the nuclear pore complex in yeast is CHMP7. The researchers found an increase in CHMP7 from the nuclei of motor cortex neurons from C9 and sporadic ALS patients, but not in neurons from the occipital cortex. The cells didn’t appear to be making additional CHMP7 but rather relocating it from the cytoplasm to the nucleus. Additional experiments revealed that CHMP7 wasn’t responding to a damaged nuclear pore but appeared to be initiating the complex’s dysfunction.
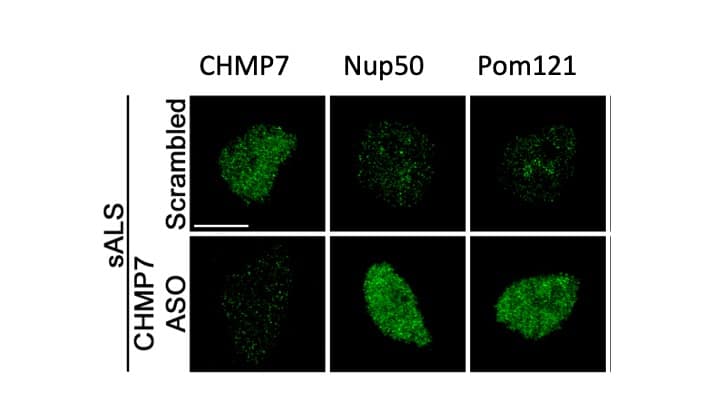
Both yeast and human cells generally export CHMP7 from the nucleus, preventing its accumulation. While this build-up could be a consequence of nuclear pore problems, the observation that CHMP7 accumulated before the subsequent Nup reduction led Coyne to hypothesize that the increase in CHMP7 actually caused the nuclear pore issues. By overexpressing a mutant version of CHMP7 that lacked a nuclear export signal (and thus wasn’t removed from the nucleus), the researchers found reduction in Nup50, POM121, and Nup133, similar to what they saw in ALS patients.
The mislocalization of CHMP7 also led to the mislocalization and loss of nuclear function of TDP43. CHMP7-triggered nuclear pore damage that led to a depletion of TDP43 from the nucleus and its subsequent accumulation in the cytoplasm. In motor neurons from ALS patients, immunostaining revealed increased CHMP7 in the nucleus even without associated TDP43 pathology, showing that the increase in CHMP7 is an early and significant event in neurodegeneration.
“Antisense oligonucleotides have emerged as a powerful and highly specific approach to treat ALS patients. They can be rapidly developed and brought to clinical experimental trials. In patient-derived cells, the ASO can reduce CHMP7 proteins and the TDP43 defect now know to be common to sporadic ALS. These observations suggest that this approach could prevent downstream neuronal dysfunction and death- in aggregate making this new “target” a potentially exciting new therapeutic opportunity,” Rothstein says.
He also points out that the study identifies one of the earliest events in not only familial ALS but also the far more common sporadic ALS. It was also carried out in a very large collection of over 30 different patient derived cell lines. As such, it acted like a modest sized in vitro clinical study.
“Before the Answer ALS program, a study like this would not have been possible. The large number of patient-derived cell lines allows us an unprecedented opportunity to understand what’s driving disease in sporadic cases. That potential is encouraging for the development of new therapeutics” Rothstein says.
Nuclear accumulation of CHMP7 precedes altered nucleocytoplasmic transport and TDP-43 mislocalization. Treatment with a CHMP7 ASO reduces levels of the protein, restores nucleocytoplasmic transport and TDP-43 localization.